A Bioconductor package to find and visualize signficantly enriched or depleted amino acid motif or amino acid group patterns in proteom dataset.
In addition to implement iceLogo in R to visualize differential amino acid sequence pattern, dagLogo can also test and visualize significant amino acid group patterns by classifying the amino acids into groups according to charge, chemistry and hydrophobicity and etc.
Ou, Jianhong, Liu, Haibo, Nirala, K N, Stukalov, Alexey, Acharya, Usha, Green, R M, Zhu, Julie L (2020). “dagLogo: An R/Bioconductor package for identifying and visualizing differential amino acid group usage in proteomics data.” Plos one, 15(11), e0242030. doi: 10.1371/journal.pone.0242030
A web application for comprehensive and efficient analyses of RNA-seq data.
OneStopRNAseq has user-friendly interfaces and offers workflows for common types of RNA-seq data analyses, such as comprehensive data-quality control, differential analysis of gene expression, exon usage, alternative splicing, transposable element expression, allele-specific gene expression quantification, and gene set enrichment analysis.
Li, R.; Hu, K.; Liu, H.; Green, M.R.; Zhu, L.J. OneStopRNAseq: A Web Application for Comprehensive and Efficient Analyses of RNA-Seq Data. Genes 2020, 11, 1165.
Nucleolus is an important structure inside the nucleus in eukaryotic cells. It is the site for transcribing rDNA into rRNA and for assembling ribosomes, aka ribosome biogenesis. In addition, nucleoli are dynamic hubs through which numerous proteins shuttle and contact specific non-rDNA genomic loci. Deep sequencing analyses of DNA associated with isolated nucleoli (NAD- seq) have shown that specific loci, termed nucleolus- associated domains (NADs) form frequent three- dimensional associations with nucleoli. NAD-seq has been used to study the biological functions of NAD and the dynamics of NAD distribution during embryonic stem cell (ESC) differentiation. NADfinder is the first software designed specifically for the bioinformatic analysis of the NAD-seq data, including baseline correction, smoothing, normalization, peak calling, and annotation.
Vertii A, Ou J, Yu J, Yan A, Liu H, Zhu LJ, Kaufman PD (2019). “Two Contrasting Classes of Nucleolus-Associated Domains in Mouse Fibroblast Heterochromatin.” Genome Research. https://genome.cshlp.org/content/29/8/1235.full.
A Bioconductor package with minimalist design for plotting elegant track layers.
This package is for the visualization of multi-omics data that can be integrated into any analysis pipeline in R. trackViewer can be used not only to visualize coverage and annotation tracks, but also to generate lollipop and dandelion plots that depict sparse and dense methylation/mutation/variant data to facilitate an integrative analysis of diverse datasets. In addition, the updated trackViewer (versions 1.19.27 and higher) has a web interface in addition to the R programming interface. Furthermore, with the ‘browseTracks’ function, users can generate interactive figures—that is, figures one can easily customize the features of by clicking, dragging, and typing.
Ou J, Zhu LJ (2019). “trackViewer: A Bioconductor package for interactive and integrative visualization of multi-omics data.” Nature Methods, 16, 453–454. doi: 10.1038/s41592-019-0430-y, https://doi.org/10.1038/s41592-019-0430-y.
A Bioconductor package for quality assessment of ATAC-seq data.
ATAC-seq (Assays for Transposase-Accessible Chromatin using sequencing) is a recently developed technique for genome-wide analysis of chromatin accessibility. Compared to earlier methods for assaying chromatin accessibility, ATAC-seq is faster and easier to perform, does not require cross-linking, has higher signal to noise ratio, and can be performed on small cell numbers. However, to ensure a successful ATAC-seq experiment, step-by-step quality assurance processes, including both wet lab quality control and in silico quality assessment, are essential. ATACseqQC package is for easily generating various diagnostic plots to help researchers quickly assess the quality of their ATAC-seq data. In addition, this package contains functions to preprocess aligned ATAC-seq data for subsequent peak calling.
Ou J, Liu H, Yu J, Kelliher MA, Castilla LH, Lawson ND, Zhu LJ (2018). “ATACseqQC: A Bioconductor package for post-alignment quality assessment of ATAC-seq data.” BMC Genomics, 19(1), 169. ISSN 1471-2164, doi: 10.1186/s12864-018-4559-3, https://doi.org/10.1186/s12864-018-4559-3.
A Bioconductor package for the visualization of motif alignment and the analysis of transcription factor binding site evolution.
This package is for the visualization of the alignment of motifs as a phylogenetic tree in different layout types. This tool facilitates the analysis of binding site diversity and conservation within families of TFs and the evolution of TFs among different species. motifStack can align DNA motifs; generate motif signatures for closely related motifs; and plot aligned motifs as a stack, a linear or a radial tree, or a word cloud of sequence logos. Different parameter settings can be used to generate diverse types of plots with color schema highlighting important data features.
This package is involved in the pipeline of finding candidate binding sites for known transcription factors via sequence matching.
Ou J, Wolfe SA, Brodsky MH, Zhu LJ (2018). “motifStack for the analysis of transcription factor binding site evolution.” Nature Methods, 15, 8-9. doi: 10.1038/nmeth.4555, http://dx.doi.org/10.1038/nmeth.4555.
A Bioconductor package for identifying off-targets with GUIDE-seq data.
The package implements GUIDE-seq analysis workflow in a flexible platform with more than 60 adjustable parameters for the analysis of datasets associated with custom nuclease applications. These parameters allow data analysis to be tailored to different nuclease platforms with different length and complexity in their guide and PAM recognition sequences or their DNA cleavage position. They also enable users to customize sequence aggregation criteria, and vary peak calling thresholds that can influence the number of potential off-target sites recovered. GUIDEseq also annotates potential off-target sites that overlap with genes based on genome annotation information, as these may be the most important off-target sites for further characterization. In addition, GUIDEseq enables the comparison and visualization of off-target site overlap between different datasets for a rapid comparison of different nuclease configurations or experimental conditions.
Zhu LJ, Lawrence M, Gupta A, Pages H, Kucukural A, Garber M, Wolfe SA (2017). “GUIDEseq: A Bioconductor package to analyze GUIDE-Seq datasets for CRISPR-Cas nucleases.” BMC Genomics, 18(1). http://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-017-3746-y.
A Bioconductor package for design of target-specific guide RNAs in CRISPR-Cas9, genome-editing systems.
The package includes functions to find potential guide RNAs for input target sequences, optionally filter guide RNAs without restriction enzyme cut site, or without paired guide RNAs, genome-wide search for off-targets, score, rank, fetch flank sequence and indicate whether the target and off-targets are located in exon region or not. Potential guide RNAs are annotated with total score of the top5 and topN off-targets, detailed topN mismatch sites, restriction enzyme cut sites, and paired guide RNAs. If GeneRfold is installed, then the minimum free energy and bracket notation of secondary structure of gRNA and gRNA backbone constant region will be included in the summary file. This package leverages Biostrings and BSgenome packages.
Zhu LJ (2015). “Overview of guide RNA design tools for CRISPR-Cas9 genome editing technology.” Front. Biol., 10(4).
Zhu LJ, Holmes BR, Aronin N and Brodsky MH (2014). “CRISPRseek: A Bioconductor Package to Identify Target-Specific Guide RNAs for CRISPR-Cas9 Genome-Editing Systems.” PLoS one, 9(9). http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4172692/.
This package uses the Naive Bayes classifier (from e1071) to assign probability values to putative polyadenylation sites (pA sites) based on training data from zebrafish. This will allow the user to separate true, biologically relevant pA sites from false, oligodT primed pA sites.
Sheppard, S., Lawson ND* and Zhu LJ*. (2013) [* denotes cocorresponding author] Accurate identification of polyadenylation sites from 3' end deep sequencing using a naïve Bayes classifier. Bioinformatics 2013
Alternative polyadenylation (APA) is one of the important post-transcriptional regulation mechanisms which occurs in most human genes. InPAS facilitates the discovery of novel APA sites from RNAseq data. It leverages cleanUpdTSeq to fine tune identified APA sites.
Search tool for RNAiCore.
ZFN target site algorithm for identifying sites for selection using the Bacterial one hybrid system.
This algorithm will also aid you in the design of libraries for the target sites using a combination of design and selection.
Please cite: https://pgfe.umassmed.edu/ZFPsearch.html.
ZFN target site algorithm for identifying sites compatible with the Lawson-Wolfe modular assembly system.
Please cite: https://pgfe.umassmed.edu/ZFPmodularsearch.html.
To create motif logo of transcription factor for preview.
Batch annotation of the peaks identified from either ChIP-seq or ChIP-chip experiments.
The package includes functions to retrieve the sequences around the peak, obtain enriched Gene Ontology (GO) terms, find the nearest gene, exon, miRNA or custom features such as most conserved elements and other transcription factor binding sites supplied by users. This package leverages the biomaRt, IRanges, Biostrings, BSgenome, GO.db, multtest and stat packages
Zhu LJ*, Gazin C, Lawson ND, Pages H, Lin SM, Lapointe DS and Green MR. (2010) [* denotes corresponding author] ChIPpeakAnno: a Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinformatics 2010, 11:237.
Analysis of high-throughput sequencing data processed by restriction enzyme digestion.
The package includes functions to build restriction enzyme cut site (RECS) map, distribute mapped sequences on the map with five different approaches, find enriched/depleted RECSs for a sample, and identify differentially enriched/depleted RECSs between samples.
Zhu LJ*, Chen PB and Fazzio TG. [* denotes corresponding author] REDseq: a Bioconductor Package for Analyzing Genome-Wide Restriction Endonuclease Accessibility Data. Bioinformatics (submitted)
Zhu LJ, Christensen RG, Kazemian M, Hull CJ, Enuameh MS, Basciotta MD, Brasefield JA, Zhu C, Asriyan Y, Lapointe DS, Sinha S, Wolfe SA and Brodsky MH. (2010) FlyFactorSurvey: a database of Drosophila transcription factor binding specificities determined using the bacterial one-hybrid system. Nucleic Acids Res., 39(Database issue): D111-D117.
The Affymetrix Human/Mouse/Rat Exon 1.0 array and Human/Mouse Exon Junction Array have been widely used for genome-wide identification of alternative splicing. Computational tools have been developed for analyzing the dataset generated from the above microaray platforms including MIDAS, MADS, PAC, Rank Product, SI/LIMMA, ARH, ANOSVA and PECA-SI. However, the existing tools are either too complicated for biologiests or lack of capablity to handle complicated experimental design. In addition, very few tools handles both exon and junction array. Here, we designed a webbased pipeline Alternative Splicing Miner (ASM) to make the analysis easy to perform yet powerful.
This tool is generated from EMBL-EBI Alternative Exon Database.
AEDB is a manual generated database for alternative exons and their properties from numerous species- the data is gathered from literature where these exons have been experimentally verified.
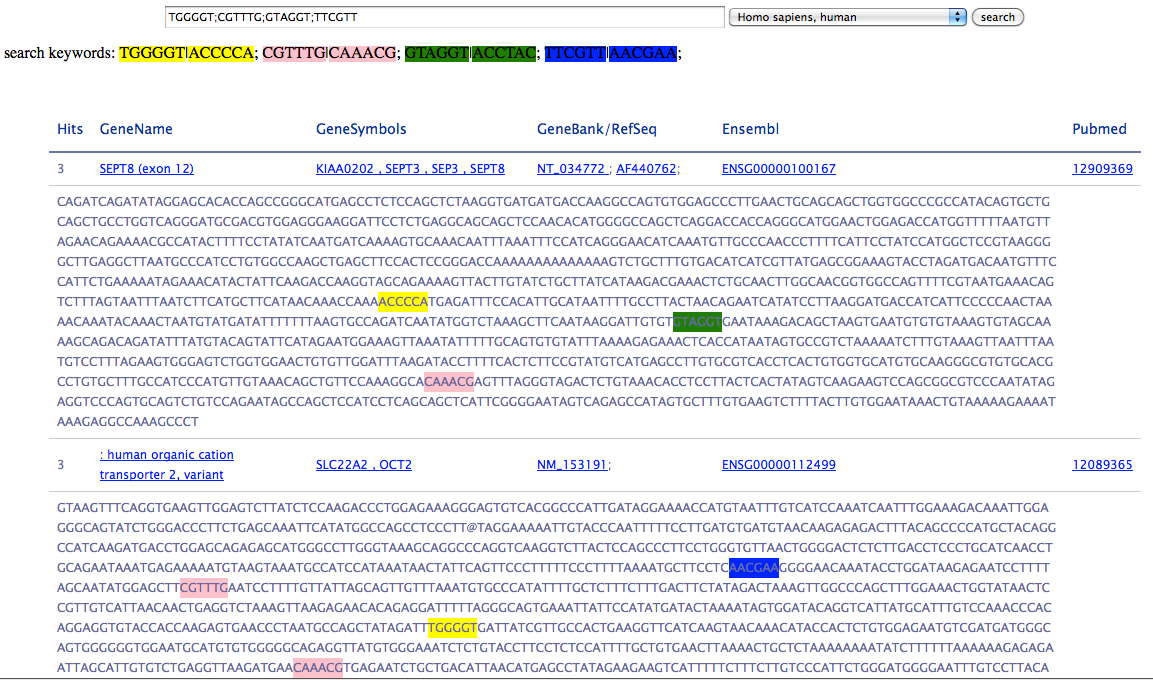
Here we extract the info from AEDB, and provied a simple seach tool by DNA pattern. Just input the DNA patterns, separate the patterns by semicolon, select species and click search button.
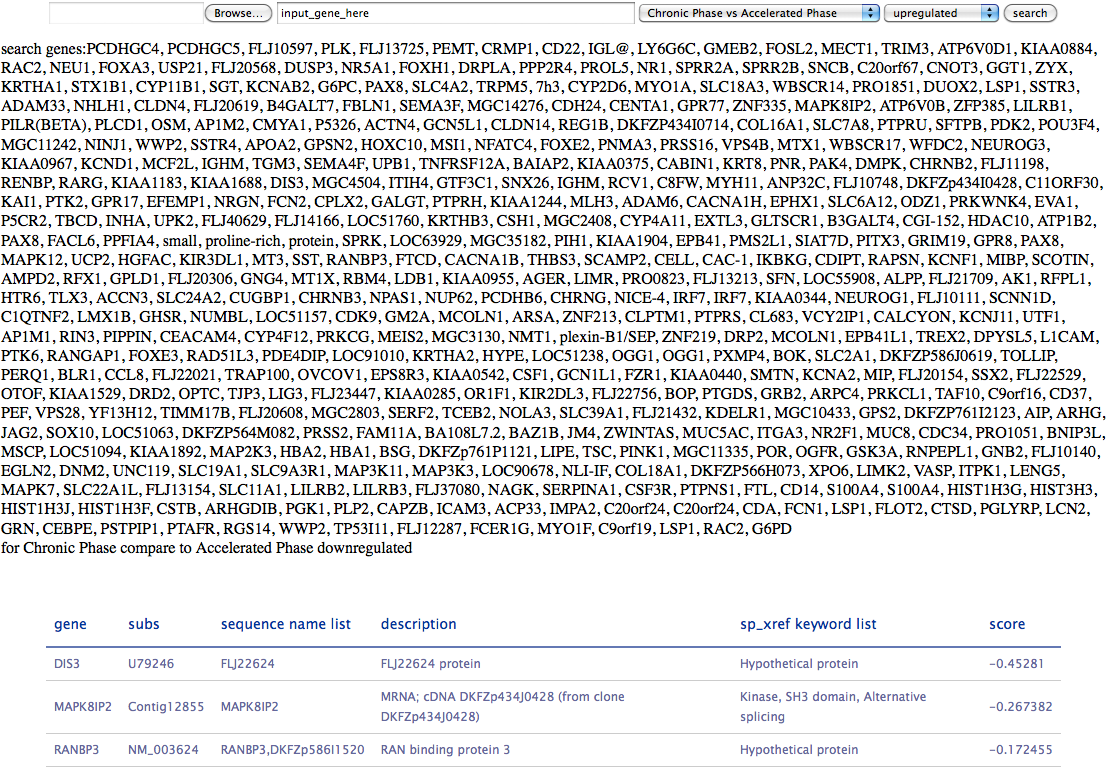
This tool is generated for gene expression profiles of cells in chronic myelogenous leukemia (CML). The data are come from three papers:
Distinct molecular phenotype of malignant CD34+ hematopoietic stem and progenitor cells in chronic myelogenous leukemia,
Chronic myelogenous leukemia molecular signature
and Molecular profiling of CD34+ cells identifies low expression of CD7, along with high expression of proteinase 3 or elastase, as predictors of longer survival in patients with CML.
You can search by a gene list (from a file or input gene names in the inputbox) and by different phase of CML.
Build Regulatory Network from ChIP-chip/ChIP-seq and Expression Data.
GeneNetworkBuilder (GNB) is a web appliation for discovering the transcriptional regulatory network for a given transcription factor (TF) of Caenorhabditis elegans, Homo sapiens and so on, using ChIP-chip (ChIP-seq) combined with gene expression profile from either RNA-seq or expression microarray experiments.
A R/Bioconductor package is also available.